На протяжении почти двух веков, прошедших с описания идиопатического паркинсонизма, данная проблема всегда оставалась в фокусе активного интереса со стороны специалистов из самых разных областей нейронаук. Это обусловлено высокой распространенностью заболевания, а также теснейшей взаимосвязью между успехами фундаментальных медико-биологических дисциплин (от патонейроморфологии до биохимии и молекулярной генетики) и прогрессом в разработке новых методов диагностики и лечения паркинсонизма, понимании тонких механизмов его развития [3, 25].
В настоящее время все случаи паркинсонизма могут быть подразделены на:
- первичный (идиопатический) паркинсонизм. К нему относятся болезнь Паркинсона и особая генетически обусловленная форма раннего паркинсонизма – так называемый ювенильный паркинсонизм;
- вторичный паркинсонизм. Данный синдром развивается в качестве одного из клинических проявлений (осложнений) ряда самостоятельных заболеваний и поражений ЦНС. Наиболее известными вариантами вторичного паркинсонизма являются сосудистый, токсический (в том числе лекарственный), посттравматический и др.;
- паркинсонизм при мультисистемных нейродегенеративных заболеваниях (так называемый паркинсонизм “плюс”). Среди заболеваний, закономерно проявляющихся синдромами паркинсонизма “плюс”, следует в первую очередь назвать прогрессирующий надъядерный паралич, множественную системную атрофию, деменцию с тельцами Леви, кортико-базальную дегенерацию;
- паркинсонизм при наследственных заболеваниях ЦНС. Это весьма обширная группа самых разных по генезу заболеваний, которая включает гепатолентикулярную дегенерацию, болезнь Галлервордена-Шпатца, дофачувствительную дистонию, ригидную форму болезни Гентингтона, ряд форм липидозов, митохондриальных энцефалопатий и т.д.
Свыше 70% всех случаев синдрома паркинсонизма в популяции приходится на долю первичного паркинсонизма и, в первую очередь, болезни Паркинсона. Заболевание встречается повсеместно, его частота варьирует от 150 до 300 на 100000 населения, резко увеличиваясь с возрастом. В возрастной группе старше 60 лет данное заболевание встречается у 1-2% лиц, что делает болезнь Паркинсона вторым по распространенности нейродегенеративным заболеванием после болезни Альцгеймера [15].
В последние годы установлено, что при постепенном, многолетнем развитии болезни Паркинсона наиболее ранние и тонкие изменения (тельца и невриты Леви) обнаруживаются в проекционных нейронах каудальных ядер мозгового ствола, и лишь затем процесс закономерно распространяется на вышележащие отделы мозга [12]. Тем не менее основные симптомы заболевания (тремор покоя, брадикинезия, мышечная ригидность, постуральные нарушения и др.) являются результатом гибели крупных дофамин-продуцирующих нейронов в компактной части черной субстанции среднего мозга, нарушения нигростриарных и других связей и недостаточности дофаминергической трансмиссии в базальных ганглиях. Благодаря механизмам нейропластичности симптоматика появляется лишь в случае гибели >70% пигментных клеток черной субстанции, что соответствует снижению уровня дофамина на 80-85% [1]. В норме в результате процессов естественного старения организма начиная с 5-го десятилетия жизни гибнет от 4,7 до 6% клеток черной субстанции в каждое десятилетие, что и определяет возрастзависимый характер болезни Паркинсона [27].
В то же время в ряде случаев паркинсонизм может развиваться и в молодом возрасте. Этому аспекту проблемы всегда уделялось гораздо меньше внимания и, более того, сложившаяся многолетняя практика почти гарантированно исключала постановку диагноза “болезнь Паркинсона” при манифестация симптомов до 40 лет. Сегодня наши подходы и сама клиническая практика кардинально изменились, так что появление различных форм первичного паркинсонизма у молодых лиц стало повседневной реальностью врача-невролога.
Интересно, что для ранних случаев паркинсонизма характерны определенные особенности чувствительности к тем или иным группам противопаркинсонических препаратов, свидетельствующие о существовании отличий в патогенезе данных форм по сравнению с “классической” болезнью Паркинсона.
Ранним паркинсонизмом (паркинсонизмом с ранним началом) принято называть случаи первичного паркинсонизма, развившегося в возрасте до 45 лет [35]. В рамках этой возрастной группы нередко выделяется самостоятельная подгруппа лиц с юношеским (ювенильным) паркинсонизмом, у которых первичный паркинсонизм манифестировал в первые два десятилетия жизни (по некоторым авторам – до 25 лет). Подавляющая часть случаев юношеского паркинсонизма связана с рецессивными мутациями недавно открытых генов паркина, DJ.1 и PINK1, продукты которых контролируют процессинг нейрональных белков и особенности окислительного метаболизма нигральных нейронов [11, 23, 41]; эти случаи обозначаются как аутосомно-рецессивный ювенильный паркинсонизм.
Наконец, в молодом возрасте могут манифестировать разнообразные паркинсоновские синдромы, обусловленные некоторыми специфическими токсинами, системными метаболическими расстройствами и другими причинами. Среди токсических паркинсоновских синдромов отметим марганцевый паркинсонизм, ставший в последние годы серьезной проблемой в связи с употреблением лицами преимущественно молодого возраста суррогатных марганецсодержащих наркотических соединений [4], а также паркинсонизм при употреблении синтетических героинов.
Таким образом, ранний паркинсонизм является чрезвычайно гетерогенным. Далее в настоящем обзоре речь пойдет о первичном паркинсонизме у лиц молодого возраста.
Несколько последних десятилетий охарактеризовались определенной тенденцией к “омоложению” паркинсонизма [6, 42]. Среди причин этого указывают:
- реализацию эффекта ряда генетических факторов;
- растущую подверженность населения развитых стран неблагоприятным эколого-средовым воздействиям;
- улучшение диагностики болезни в ее начальных стадиях, связанное с общим технологическим прогрессом в клинической медицине.
Все эти факторы заслуживают детального рассмотрения.
Возможность развития “истинной” болезни Паркинсона в молодом возрасте подтверждается обнаружением на секции типичной “Леви-патологии” (в том числе с использованием иммуногистохимической окраски на a-синуклеин) в соответствующих отделах головного мозга у лиц, у которых симптоматика асимметричного леводопачувствительного паркинсонизма впервые появилась на 4-5-м десятилетии жизни [46]. Эти случаи ранней болезни Паркинсона могут носить как спорадический, так и семейный характер. Раннее начало болезни Паркинсона принято связывать, в первую очередь, с генетическими факторами, многие из которых раскрыты и достаточно хорошо изучены благодаря интенсивному прогрессу последних лет в области молекулярной генетики. Показана ассоциация болезни Паркинсона с рядом полиморфизмов в генах детоксикации ксенобиотиков, системы антиоксидантной защиты клетки, транспорта и метаболизма дофамина, липидного обмена, митохондриального цикла. Гены предрасположенности к болезни Паркинсона представлены ниже:
Гены систем клеточной детоксикации и антиоксидантной защиты
- параоксоназа-1
- убиквитин-С-концевая гидролаза L1
- цитохром Р450 (CYP2D6)
- N-ацетилтрансфераза-2
- семейство ферментов глутатин-трансферазы гемоксигеназа-1
- ферменты a-кетоглутарат-дегидрогеназного комплекса
- супероксид-дисмутаза
Гены транспорта и метаболизма дофамина
- моноаминоксидазы А и B
- катехол-О-метилтрансфераза
- тирозингидроксилаза
- транспортеры дофамина
- дофаминовые рецепторы D2, D3, D4 и D5
Митохондриальный геном
- тРНКГлу
- митохондриальная ДНК (отдельные полиморфизмы)
- комплекс I электронной дыхательной цепи
Другие гены
- NO-синтазы (nNOS, iNOS)
- аполипопротеин Е
- нейротрофические факторы
Носительство неблагоприятных аллельных вариантов данных генов достоверно повышает риск заболевания, т.е. формирует генетическую предрасположенность к болезни Паркинсона, которую можно оценить количественно – например, путем стандартного расчета соотношения шансов (показатель OR) [39]. Нами и другими исследователями установлено, что выявленные ассоциации (как и семейная кластеризация болезни) значительно выше в группе паркинсонизма молодого возраста [8, 16, 36]. Более того, комбинация нескольких неблагоприятных полиморфизмов усиливает предрасположенность к болезни Паркинсона и ведет к более ранней манифестации симптомов (аддитивный эффект “генов риска”) [16]. Таким образом, соотношение генетических и средовых факторов в развития болезни Паркинсона неодинаково в различных возрастных группах больных (рисунок): в молодой группе пациентов наиболее значим удельный вес генетической составляющей, тогда как у пожилых пациентов роль генетики становится менее четкой, и на первый план выходят средовые и иные факторы.
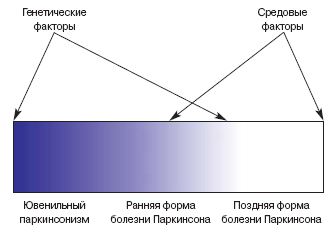
Взаимодействие генетических и средовых факторов при различных формах первичного паркинсонизма. Темная часть спектра отражает удельный вес генетических факторов, светлая – средовых.
Говоря о генетическом грузе, нельзя забывать и о накоплении мутаций митохондриальной ДНК, скорость которого в современных условиях может быть более высокой, а последствия в виде нарушения энергообеспечения нейронов – более драматичными и ранними [22]. Отметим в этой связи, что более чем у половины лиц с болезнью Паркинсона отмечается снижение активности комплекса I митохондрий в различных тканях, в том числе в мозге [38].
Известным модификатором возраста начала болезни Паркинсона и ряда других нейродегенеративных заболеваний является генетический полиморфизм аполипопротеина Е (ароЕ) – белка, имеющего отношение к репарации клеточных мембран и мобилизации “строительного” холестерина [14, 26, 47]. Как в нашей работе, так и в ряде работ зарубежных авторов было установлено, что наличие аллеля ароЕ-e4 способствует более раннему началу болезни Паркинсона [2, 47].
В наибольшей степени этот эффект проявляется при гомозиготности по данному варианту ароЕ (генотип e4/e4): в таких случаях, согласно нашим наблюдениям, возраст начала болезни составил 35-37 лет.
По разным данным, от 5 до 10% всех случаев болезни Паркинсона имеют не мультифакторную, а моногенную природу, представляя собой заболевания с аутосомно.доминантным наследованием [43]. Они обусловлены передачей в поколениях патогенетически значимых мутаций в генах a-синуклеина, UCH.L1, LRRK2 и некоторых других. Последний, совсем недавно открытый ген (LRRK2) имеет особое значение, поскольку он может обусловливать до 1% всех случаев болезни Паркинсона в популяции – в том числе (поскольку пенетрантность гена не превышает 70%) при отсутствии четкого семейного анамнеза [18].
Можно заключить, что в общем ряду случаев “обычной” спорадической болезни Паркинсона нередко “маскируются” те или иные наследственные формы, что подчеркивает гетерогенность данной патологии. Для наследственных случаев болезни Паркинсона характерно относительно раннее появление симптомов: так, в серии на ших случаев заболевание у носителей мутации в гене LRRK2 могло проявиться уже в 39 лет, описаны и более ранние варианты LRRK2.формы болезни Паркинсона.
В ряду факторов, способствующих развитию болезни Паркинсона, большое значение придается подверженности населения развитых стран мира неблагоприятным эколого.средовым воздействиям и, в первую очередь, потенциальным нейротоксинам [10, 42]. Наиболее вероятными кандидатами на роль экзогенных “каузативных” токсинов при болезни Паркинсона являются некоторые пестициды: показано, что в условиях in vitro пестициды способны провоцировать конформационные изменения молекулы a-синуклеина (это ключевой этап патогенеза болезни Паркинсона) и ускорять формирование патологических включений в нейронах [40]. Хроническое системное воздействие пестицидов воспроизводит в эксперименте клинические характеристики болезни Паркинсона [9]. Эпидемиологические исследования подтверждают эти заключения. Так, распространенность болезни Паркинсона в сельских популяциях среди фермеров оказалась почти в 1,4 раза выше по сравнению с городским населением [24], а риск болезни Паркинсона у работников плантаций – в 1,5-2 раза выше общепопуляционного [33], что может служить подтверждением роли в развитии заболевания пестицидов и других агентов, контакт с которыми по определению выше у работников, занятых в сельском хозяйстве. Предполагается, что агрессивное агрохимическое производство и соответствующая экологическая обстановка, характерные для ряда регионов мира, могут явиться серьезным фактором, способствующим общему росту заболеваемости болезнью Паркинсона и, при особенно неблагоприятном развитии событий, накоплению более ранних случаев болезни в определенных субпопуляциях. Вероятность манифестации раннего паркинсонизма в такой ситуации особенно возрастает у лиц – носителей неблагоприятных аллельных вариантов “предрасполагающих” генов, отвечающих в организме за процессы клеточной детоксикации (см. выше) [42]. Интересно, что ассоциации болезни Паркинсона с аллельными вариантами генов.детоксикантов (CYP2D6, GSTP1 и др.) особенно значимы именно в в группе лиц, имевших контакт с пестицидами [17, 28]. Этот пример наглядно иллюстрирует взаимодействие генетических и средовых факторов в развитии болезни Паркинсона.
Определенная роль в увеличении доли молодых пациентов с болезнью Паркинсона принадлежит совершенствованию методов диагностики и улучшению распознавания наиболее ранних случаев болезни (и даже “предболезни”). Отметим, во-первых, внедрение в практику понятия группа риска, к которой принадлежат, в частности, ближайшие родственники пациентов, имеющие в несколько раз более высокую вероятность развития болезни Паркинсона по сравнению с общей популяцией [43]. Именно в этой группе лиц, хорошо осведомленных об имеющейся семейной отягощенности, становится возможным тщательный мониторинг, ориентированный на выявление максимально ранних нарушений в двигательной сфере. Во-вторых, определенное значение имела разработка новейших методов нейровизуализации (КТ и МРТ, ОФЭКТ, ПЭТ), позволяющих осуществлять более точный дифференциальный диагноз и объективизировать тонкие нарушения дофаминового обмена в базальных ганглиях [13, 34]. Наконец, нельзя не отметить роль разработанных международным сообществом унифицированных критериев клинической диагностики болезни Паркинсона, позволивших усовершенствовать и стандартизировать подходы к раннему выявлению и постановке диагноза данного заболевания.
На приведенном ранее рисунке в левой части спектра представлено особое генетически детерминированное заболевание – аутосомно-рецессивный ювенильный паркинсонизм (АР.ЮП), который обусловливает значительную часть случаев “молодого” паркинсонизма и характеризуется рядом своеобразных клинико-морфологических проявлений [2, 23]. Данное заболевание встречается практически во всех изученных популяциях мира.
Основное значение в развитии АР-ЮП имеет ген, локализованный на хромосоме 6q и кодирующий новый белок с убиквитин-лигазной функцией – паркин [23]. Показано, что паркин является важнейшим звеном системы клеточной защиты и, в частности, непосредственно участвует в деградации a-синуклеина – классического белкового маркера болезни Паркинсона в составе характерных интранейрональных включений (телец Леви) [37].
Морфологическая картина АР.ЮП характеризуется гибелью нейронов и глиозом в компактной части черной субстанции и голубоватом пятне, отличаясь от “классической” болезни Паркинсона отсутствием телец Леви в дегенерирующих нейронах [23].
Дебют симптомов АР-ЮП чаще всего приходится на 2-3.е десятилетие жизни, первым проявлением заболевания может быть постепенно развивающийся синдром паркинсонизма либо дистония стоп. Для синдрома паркинсонизма в развернутой стадии АР.ЮП типично сочетание брадикинезии, мышечной ригидности, постуральных нарушений с пирамидными симптомами, а также нередкое отсутствие стадии гемипаркинсонизма.
Особенностью тремора при АР-ЮП является его статокинетический характер, который может сочетаться с типичным паркинсоновским тремором покоя. Проявления дистонии при АР-ЮП, появившись у ряда пациентов в дебюте болезни, могут сохраняться на протяжении многих лет. Важной особенностью болезни, имеющей существенное дифференциально-диагностическое значение, является весьма раннее появление разнообразных и нередко сложных по своей структуре леводопа-индуцированных дискинезий, которые могут возникнуть уже при приеме чрезвычайно низких доз препарата (30-70 мг леводопы). Еще одной отличительной чертой АР-ЮП, отмечаемой у большинства больных, является наличие флуктуаций в выраженности симптомов паркинсонизма и дистонии на протяжении дня: наилучшее состояние отмечается утром или после дневного сна, а к вечеру тяжесть клинических проявлений нарастает. Это сближает клинику АР-ЮП с проявлениями дофачувствительной торсионной дистонии.
Гомозиготные мутации гена паркина при первичном паркинсонизме с началом болезни до 20 лет выявляются более чем в 70% семейных и в 15% спорадических случаев [30]. Заметно реже при АР-ЮП выявляются гомозиготные мутации в генах PINK1 и DJ-1 [21, 41]. Изредка у гомозиготных носителей мутаций гена паркина описывается начало болезни в более позднем возрасте (вплоть до 6-го десятилетия жизни!), и такие случаи могут быть неотличимы от “классической” болезни Паркинсона [30]. Это демонстрирует определенную условность термина “ювенильный паркинсонизм” применительно к паркин-ассоциированным формам патологии (для обозначения этих синдромов более адекватным представляется получающий всё большее признание термин “паркинопатии”).
Совсем недавно появились основания предполагать, что гетерозиготное носительство мутации в гене паркине иногда достаточно для развития доминантной формы паркинсонизма – наиболее вероятно, в результате падения ниже критического “порога” лигазной активности белкового продукта гена (механизм гаплонедостаточности) [44]. Прижизненное ПЭТ-исследование показало отчетливое снижение захвата флюородопы в стриатуме у лиц, имеющих мутантный и нормальный аллель паркина, что является четким нейровизуализационным свидетельством дофаминергической дисфункции у гетерозигот [20].
С учетом этих данных нами был проведен поиск структурных перестроек в гене паркине у 107 пациентов с ранним паркинсонизмом (возраст начала до 45 лет). При этом мутации паркина были выявлены у 13,1% больных (14 пациентов), в том числе в 12 случаях из 14 – гетерозиготные делеции и дупликации. Аналогичные данные получены и другими исследователями [30, 32, 44].
Таким образом, гаплонедостаточность по паркину действительно может определять гибель дофаминовых нейронов и служить значимым фактором риска первичного паркинсонизма в молодом возрасте. Подчеркнем, что заболевание у гетерозиготных носителей паркин.мутаций отличается от АР-ЮП по своим генетическим и клиническим характеристикам, являясь самостоятельным и пока еще мало изученным вариантом паркин-ассоциированного паркинсонизма.
Раннее начало первичного паркинсонизма предъявляет повышенные требования к рационализации назначаемой терапии, поскольку такая терапия должна быть ориентирована на длительную перспективу, в идеале – на десятилетия вперед.
Большинство исследователей предпочитает начинать лечение раннего паркинсонизма с агонистов дофаминовых рецепторов (АДР) [5, 27, 29]. Целесообразность их использования в данной группе пациентов обусловлена лучшей переносимостью АДР у молодых по сравнению с пожилыми больными, а также необходимостью принимать во внимание возможность быстрого появления выраженных дискинезий у молодых пациентов в случае назначение леводопы (это наиболее типично для различных вариантов ювенильного паркинсонизма). Кроме того, именно для пациентов молодого возраста в начальной стадии болезни важен оказываемый препаратами из группы АДР нейропротекторный эффект (подтвержденный в эксперименте и в некоторых клиниконейровизуализационных исследованиях) [31, 45], что позволяет в определенной степени пролонгировать течение болезни. АДР-опосредованную нейропротекцию связывают с уменьшением синаптического кругооборота дофамина, стимуляцией D1-рецепторов, синтезом белков с антиоксидантными свойствами, стимуляцией аутотрофической активности нейронов, уменьшением секреции эксайтотоксина глутамата.
При необходимости у пациентов молодого возраста АДР можно комбинировать с ингибиторами МАО-В, а также амантадинами (мидантан, ПК-Мерц и др.). Последняя группа препаратов достаточно перспективна для лечения именно молодых случаев паркинсонизма, поскольку имеющиеся данные свидетельствуют о наличии у них свойств антагонистов NMDA-рецепторов глутамата [19]; таким образом, амантадины способны реализовывать свое предполагаемое нейропротекторное действие на уровне “эксайтотоксического каскада”.
В молодой возрастной группе для борьбы с тремором (весьма резистентным к терапии симптомом болезни Паркинсона) более свободно могут быть назначены центральные холинолитики, обычно не рекомендуемые у пожилых лиц в связи с большим числом общесоматических противопоказаний, опасностью нарастания когнитивных нарушений и риском развития психотических состояний на фоне атрофии мозга. Следует помнить, что у молодых больных центральные холинолитики назначаются в минимально возможной дозировке, а общая продолжительность непрерывного лечения данными препаратами не должна превышать 3-5 лет.
На определенном этапе болезни при нарастании двигательных нарушений возникает необходимость приема препаратов леводопы. Согласно современным представлениям, “опасность” леводопы с точки зрения ее неблагоприятного влияния на течение болезни Паркинсона остается недоказанной, а несвоевременное (чрезмерно отсроченное) назначение леводопы может нивелировать имеющийся терапевтический потенциал заместительной терапии и, тем самым, оказать негативное влияние на прогноз болезни и качество жизни [5, 7]. Избегая необоснованной “леводопофобии”, следует помнить, что у молодых пациентов требуется особенно тщательное “титрование” разовых и суточных доз леводопы, минимизирующее проявления нередко весьма мучительных для больного дискинезий.
Контроль двигательных осложнений леводопа-терапии предполагает назначение разнообразных патогенетических и симптоматических корректоров (АДР, ингибиторы КОМТ и новый комбинированный препарат леводопы Сталево – см. далее, бензодиазепины и др.).
Согласно нашему опыту, не менее чем у половины пациентов с ювенильным паркинсонизмом применение леводопасодержащих препаратов было невозможным без их сочетания с АДР. С учетом ожидаемой продолжительности жизни и необходимости максимального отсрочивания двигательных флуктуаций, у молодых больных стратегически обоснованным представляется начало терапии леводопой с ее пролонгированных форм.
В последние годы появился ряд новых направлений лечения паркинсонизма, связанных как с функциональной нейрохирургией (высокочастотная электростимуляция базальных ганглиев), так и с оригинальными методами трансдермальной доставки противопаркинсонических средств и технологиями малоинвазивной хирургии. Заслуживает внимания так называемая концепция постоянной дофаминергической стимуляции, которая может быть реализована, в частности, посредством дозируемого введения леводопы через постоянную дуоденальную помпу; это позволяет эффективно купировать тяжелые двигательные флуктуации у пациентов в развернутой стадии болезни. Еще один подход, позволяющий пролонгировать и “физиологизировать” эффект леводопы, изящно реализован в новой и чрезвычайно перспективной лекарственной форме, представляющей собой комбинацию леводопы, карбидопы и энтакапона (препарат Сталево). Имеющийся к настоящему времени опыт показывает высокую эффективность Сталево в уменьшении выраженности двигательных флуктуаций и снижении риска развития дискинезий как в клинике, так и в эксперименте, что связывают с улучшенной фармакокинетикой леводопы (одновременное ингибирование периферической дофа-декарбоксилазы и катехол-О-метилтрансферазы). Обсуждается потенциальный протективный эффект Сталево при раннем назначении препарата.
Использование вышеуказанных и ряда других новых технологий при различных формах первичного паркинсонизма показано, в первую очередь, у молодых пациентов, имеющих меньшее число противопоказаний и неблагоприятных прогностических факторов. Это существенно расширяет имеющиеся возможности эффективной помощи людям трудоспособного возраста, страдающим ранним паркинсонизмом, что имеет не только медицинский, но и несомненный социально-экономический эффект.
Литература
- Бархатова В.П. Нейротрансмиттеры и экстрапирамидная патология. М., 1988.
- Загоровская Т.Б. и др. // Журн. неврологии и психиатрии им. С.С. Корсакова. 2004. № 8. C. 66.
- Иллариошкин С.Н. Конформационные болезни мозга. М., 2003.
- Исмаилова Т.Ф. Особенности клинических проявлений токсической энцефалопатии, вызванной употреблением суррогатных психоактивных веществ, содержащих марганец: Автореф. дис. … канд. мед. наук. М., 2005.
- Левин О.С. // Атмосфера. Нервные болезни. 2005. № 1. С. 10.
- Пчелина С.Н. и др. // Мед. генетика. 2003. № 9. С. 411.
- Agid Y. et al. // Lancet. 1998. V. 351. P. 851.
- Akhmedova (Pchelina) S. et al. // J. Neurol. Sci. 2001. V. 184. P. 179.
- Betarbet R. et al. // Nat. Neurosci. 2000. V. 3. P. 1301.
- Bonifati V. // 4th International Symposium on Parkinson’s disease and restless legs syndrome. Stresa, 2005. P. 42.44.
- Bonifati V. et al. // Science. 2003. V. 299. P. 256.
- Braak H. et al. // Neurobiol. Aging. 2003. V. 24. P. 197.
- Brooks D.J. // Parkinson’s Disease and Movement Disorders / Ed. by Jancovic J., Tolosa E. Baltimore, 1998. P. 991.
- Chapman J. et al. // Neurology. 2001. V. 57. P. 1482.
- De Rijk M.C. et al. // Neurology. 1995. V. 45. P. 2143.
- Djuric G., Illarioshkin S.N. et al. // XII kon. gres neurologa Srbije i Crne Gore sa medunarodnim ucesce. Novi Sad, 2004. P. 45.46.
- Elbaz A. et al. // Ann. Neurol. 2004. V. 55. P. 430.
- Foroud T. // Neurology. 2005. V. 65. P. 664.
- Greenamyre J.T. // Ann. Neurol. 1996. V. 39. P. 537.
- Hilker R. et al. // Neurosci. Lett. 2002. V. 323. P. 50.
- Ibanez P. et al. // Neurology. 2003. V. 61. P. 1429.
- Johns D.R. // N. Engl. J. Med. 1995. V. 333. P. 638.
- Kitada T. et al. // Nature. 1998. V. 392. P. 605.
- Lai E.C., Moore S. // Neurology. 2006. V. 66. Suppl. 2. A213.
- Lansbury P.T., Brice A. // Curr. Opin. Cell Biol. 2002. V. 14. P. 653.
- Mahley R. et al. // Ann. N.Y. Acad. Sci. 1996. V. 777. P. 139.
- Marsden C.D. // J. Neurol. Neurosurg. Psychiatry. 1994. V. 57. P. 672.
- Menegon A. et al. // Lancet. 1998. V. 352. P. 1344.
- Miyasaki J.M. et al. // Neurology. 2002. V. 58. P. 11.
- Morrison K.E. // Brain. 2003. V. 126. P. 1250.
- Parkinson Study Group // JAMA. 2002. V. 287. P. 1653.
- Periquet M. et al. // Brain. 2003. V. 126. P. 1271.
- Petrovitch H. et al. // Arch. Neurol. 2002. V. 59. P. 1787.
- Piccini P. et al. // Ann. Neurol. 1997. V. 41. P. 222.
- Quinn N. et al. // Mov. Disord. 1987. V. 2. P. 73.
- Rocca W.A. et al. // Ann. Neurol. 2004. V. 56. P. 495.
- Shimura H. et al. // Nat. Genet. 2000. V. 25. P. 302.
- Swerdlow R.H. et al. // Ann. Neurol. 1996. V. 40. P. 663.
- Tan E.K. et al. // Neurology. 2000. V. 55. P. 533.
- Uversky V.N. et al. // FEBS Lett. 2001. V. 500. P. 105.
- Valente E.M. et al. // Science. 2004. V. 304. P. 1158.
- Veldman B. et al. // Clin. Neurol. Neurosurg. 1998. V. 100. P. 15.
- Vila M., Przedborski S. // Nat. Med. 2004. V. 10. Suppl. S58.
- West A. et al. // Amer. J. Med. Genet. 2002. V. 114. P. 584.
- Whone A.L. et al. // Ann. Neurol. 2003. V. 54. P. 93.
- Wszolek Z.K. et al. // Neurology. 2004. V. 62. P. 1619.
- Zareparsi S. еt al. // Amer. J. Med. Gen. 2002. V. 107. P. 156.
Автор: Иллариошкин С.Н.
Источник: Атмосфера. Нервные болезни, 3, 2006